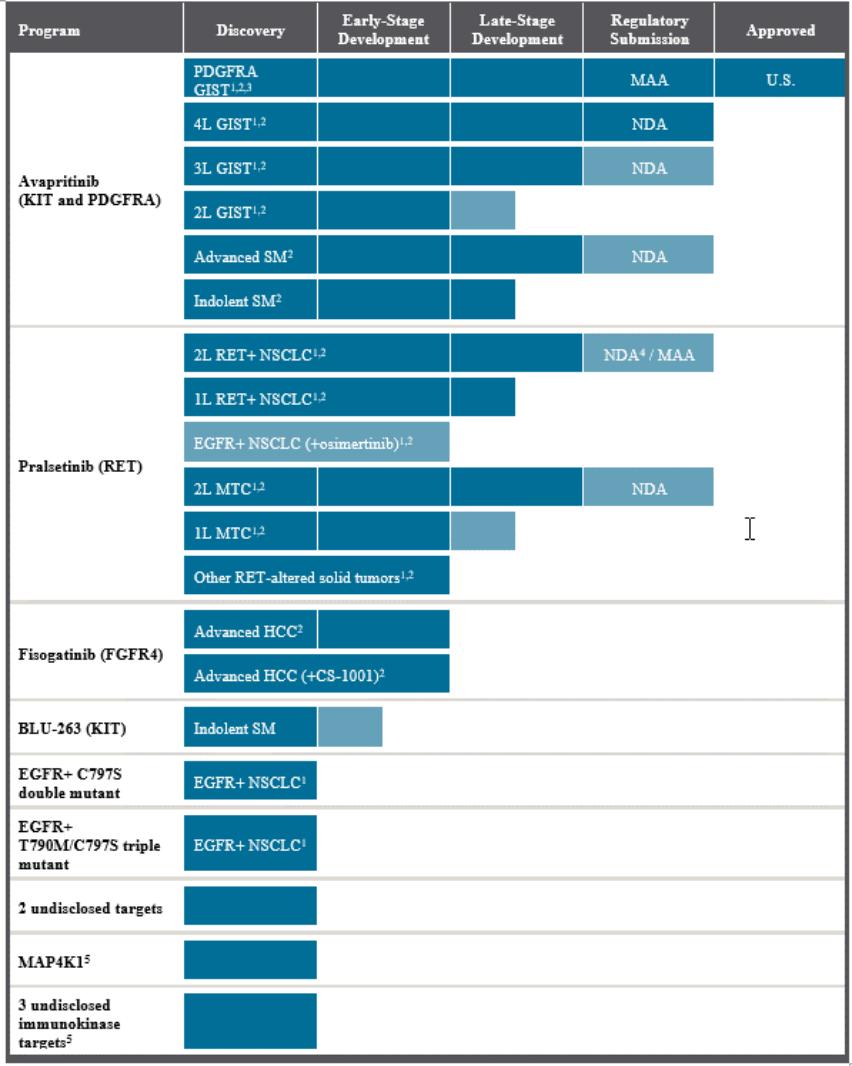
截至数据截止日期,报告了其他RET改变的癌症患者的临床活动数据:
●应用RECIST 1.1版对6例PTC患者进行疗效评估。在这些患者中,ORR阳性率为83%(3例确诊PRs,2例PRs有待证实)。
●5例PTC患者持续治疗1年以上,8例PTC患者在数据截止日期仍在接受治疗。
●在其他RET融合癌(包括胰腺癌(一例确诊PR,一例PR尚待确认)和肝内胆管癌(一例确诊PR)的患者中观察到额外的反应。
4例患者(其中2例为RET融合型NSCLC,2例为RET-突变型MTC)曾接受过Selpercatinib治疗。其中:
●两名病人进行了公关,其中一名在数据截止日期得到确认,一名在数据截止日期待决,随后在提交前确认。
●1例患者有稳定的疾病与X线肿瘤缩小,并继续治疗,直到数据截止日期。
安全数据。截止数据截止日期,226例患者开始剂量为400 mg/d,安全性评价。在所有患者中,普拉西替尼耐受性良好,研究者报告的大多数AES为1级或2级。在所有级别中,调查人员报告的最常见的治疗方法是便秘、高血压、天冬氨酸转氨酶升高、中性粒细胞减少、腹泻、疲劳、贫血、丙氨酸转氨酶升高和血肌酐升高(≥15%)。在所有级别中,最常见的紧急治疗是便秘、高血压、天冬氨酸转氨酶升高、中性粒细胞减少、腹泻、疲劳、贫血。研究人员报告3或4级治疗相关的AES(≥2%)包括中性粒细胞减少、高血压、贫血、血肌酸磷酸激酶升高和白细胞减少。
在所有患者中,只有4%的患者因与治疗相关的不良反应而停止使用普拉西替尼治疗。7%的RET融合的NSCLC患者因治疗相关的AEs而停止使用pralsetinib治疗,而没有任何RET突变的MTC患者因治疗相关的AEs而停止使用pralsetinib的治疗。
2020年1月非小细胞肺癌患者普拉西替尼的第一线数据
报告了使用pralsetinib治疗的患者的一线疗效数据,这些患者可根据RECIST 1.1版评估疗效,这是由盲目的独立中央审查确定的。,截止日期为2019年11月18日。所有患者均接受400 mg qd的拟议剂量。
在80例RET融合阳性NSCLC患者中,以铂为基础的化疗治疗后,ORR为61%(95%可信区间,或CI:50-72%)。总的来说,95%的患者有肿瘤缩小,其中14%的患者有完全消退的目标肿瘤。DOR中位数未达到(95%CI:11.3个月,不可估计)。
26例接受非小细胞肺癌治疗的非小细胞肺癌患者的ORR为73%(95%CI:52-88%)(所有反应均得到证实),12%的患者达到CR。所有患者均有肿瘤缩小。
上线安全数据与2019年6月ASCO年会报告的数据一致。Pralsetinib耐受性良好,大多数AES为1级或2级。在所有参加ART试验的患者中,以400 mg qd(N=354)的拟议剂量治疗的所有患者中,只有4%的患者因与治疗相关的AEs而停止使用pralsetinib治疗。